
When you pick up a generic pill at the pharmacy, you expect it to work just like the brand-name version. But how does the FDA make sure it does? The answer lies in a quiet, lab-based process called dissolution testing. It’s not glamorous. No patients are involved. No clinical trials are run. Yet, this single test is one of the most powerful tools the FDA uses to guarantee that every generic drug you take is safe, effective, and identical in performance to its brand-name counterpart.
Why Dissolution Testing Matters
Dissolution testing measures how quickly a drug releases its active ingredient into a simulated liquid environment - think of it as mimicking what happens in your stomach and intestines. For a generic drug to be approved, it must release its medicine at the same rate and to the same extent as the original product. If it releases too slowly, you won’t get enough medicine into your bloodstream. If it releases too fast, you could overdose or experience side effects. The FDA doesn’t just ask manufacturers to guess at this. They demand hard data. For oral solid doses - tablets and capsules - every single generic must pass this test before it hits the shelf. The goal? To replace human bioequivalence studies with reliable lab tests. Why? Because testing on people is expensive, time-consuming, and ethically complex. Dissolution testing gives the FDA a way to predict how a drug will behave in the body without ever needing a volunteer.How the FDA Sets the Rules
The FDA doesn’t use one-size-fits-all standards. Each drug has its own dissolution profile, and the method must match its chemistry. For example, a simple aspirin tablet is very different from a 24-hour pain reliever with a special coating. According to the FDA’s 2023 guidance, manufacturers must submit five key types of data:- How soluble the active ingredient is in water and digestive fluids
- Exactly how the test is set up - including the type of machine (usually USP Apparatus 1 or 2), rotation speed (often 50-100 rpm), fluid volume (500-900 mL), and pH level
- Proof that the test works under small changes - like a slightly warmer temperature or a different buffer
- Validation that the lab can accurately measure how much drug is dissolved
- Demonstration that the test can tell the difference between good and bad formulations - especially for low-solubility drugs
For immediate-release tablets (the kind you swallow and expect to work within 30 minutes), the standard is clear: at least 80% of the drug must dissolve within 45 minutes. But for some high-solubility drugs - like those classified under the Biopharmaceutics Classification System (BCS) Class I - the FDA allows a simpler test. If the drug dissolves quickly in 0.1N HCl (a stomach-like acid), a single measurement at 30 minutes using 900 mL of fluid is enough. This rule, from the FDA’s 2018 guidance, cuts development time for generics by months.
The f2 Similarity Factor: The Gold Standard
Comparing two dissolution curves isn’t just about eyeballing graphs. The FDA uses a mathematical tool called the f2 similarity factor. This score, ranging from 0 to 100, tells you how closely the generic matches the brand-name drug over time. An f2 score of 50 or higher means the two products are statistically equivalent. A score below 50 raises red flags. For example, if the brand-name drug releases 70% of its drug at 30 minutes, and the generic only releases 45%, the f2 score might be 32. That’s a rejection. The manufacturer must go back, tweak the formulation, and retest. This isn’t theoretical. In 2023, over 2,800 drug products had FDA-recommended dissolution methods listed in their public database. These aren’t suggestions - they’re benchmarks. Manufacturers who ignore them risk having their application denied.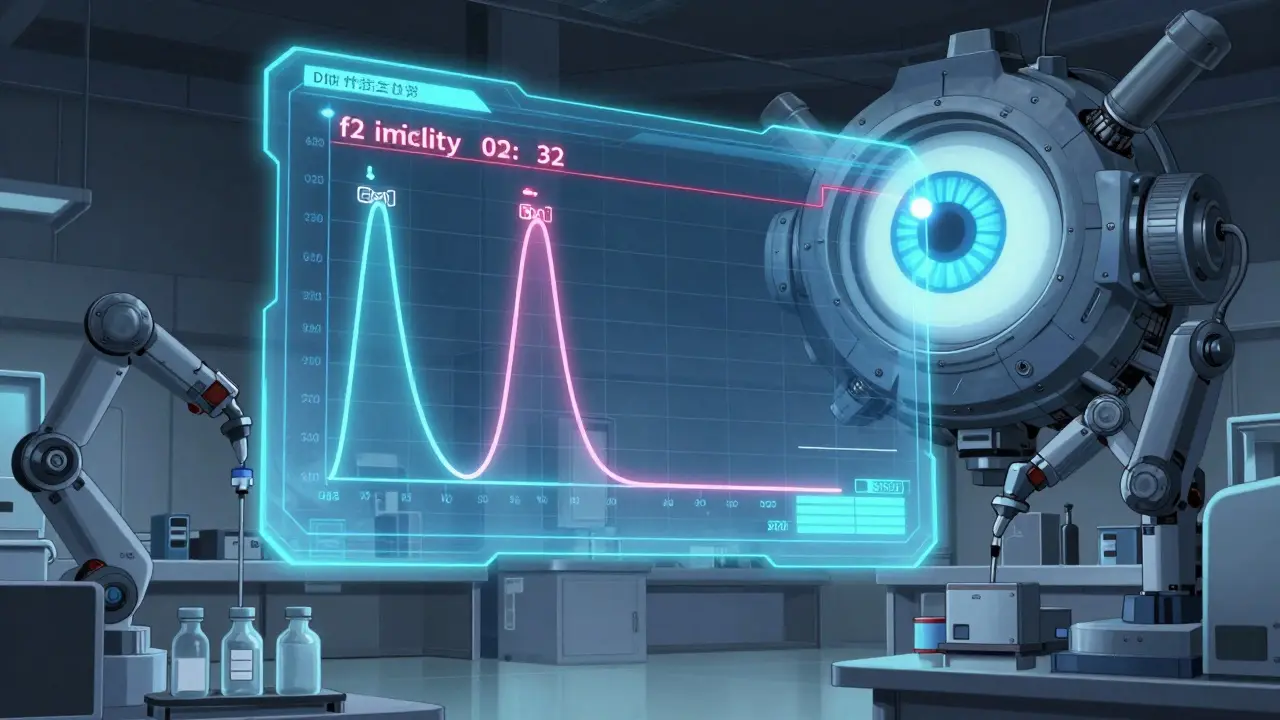
Special Cases: Modified-Release and Low-Solubility Drugs
Not all drugs are created equal. Extended-release pills, like those for ADHD or high blood pressure, are designed to release medicine slowly over hours. Testing these is far more complex. For these products, the FDA requires testing under multiple pH conditions - typically pH 1.2 (stomach), pH 4.5 (upper intestine), and pH 6.8 (lower intestine). Why? Because the drug’s release must be consistent no matter where it is in your gut. If a pill dissolves too fast in acidic conditions, it could dump its entire dose at once - a dangerous phenomenon called “dose-dumping.” To catch this, the FDA also requires alcohol challenge tests. Many people take their meds with a glass of wine or beer. If a drug releases 40% more when mixed with 40% ethanol, that’s a safety hazard. The FDA has rejected multiple generic versions of extended-release opioids and stimulants for failing this test. Low-solubility drugs - like many antifungals or cholesterol-lowering agents - are another challenge. These drugs don’t dissolve easily. A poor dissolution method might not detect differences between formulations that behave very differently in the body. That’s why the FDA demands “discriminatory ability” in the test. The method must be sensitive enough to catch small changes in excipients, particle size, or coating.What Happens After Approval?
Approval doesn’t mean the testing stops. The FDA’s SUPAC-IR guidelines require manufacturers to prove that any change - whether it’s a new factory, a different supplier of raw material, or even a slight tweak in the mixing process - doesn’t alter the dissolution profile. If a company switches from one tablet press to another, they must retest. If they change the binder from cornstarch to cellulose, they must retest. This isn’t bureaucracy - it’s consistency. A change that seems minor to a manufacturer can have major effects on how the drug works in your body. The FDA also uses dissolution testing to monitor quality after a drug is on the market. Random samples from pharmacies are tested. If a batch fails, the FDA can issue a recall - even if no patient has been harmed.Why This System Works
The brilliance of dissolution testing is its efficiency. For BCS Class I drugs, the FDA can approve a generic without ever running a single human bioequivalence study. That means faster access to affordable medicine. In 2023, nearly 35% of generic approvals used this streamlined path - up from 25% in 2020. But this system only works because it’s grounded in science. The FDA doesn’t just copy methods from the USP (United States Pharmacopeia). They require manufacturers to prove their method works for that specific drug. They demand evidence. They require transparency. And they don’t accept shortcuts. The result? Generic drugs that work just as well as brand-name ones - often at a fraction of the cost. Over 90% of prescriptions in the U.S. are filled with generics. That’s possible because of dissolution testing.
What Manufacturers Face
Developing a dissolution method isn’t easy. For complex products, it can take 6 to 12 months of lab work. A single ANDA application often includes 50 to 100 pages of dissolution data - method validation, stability tests, comparative curves, statistical analysis. And the FDA doesn’t always approve the first submission. If the data is incomplete, the agency issues a “complete response letter,” asking for more. This delays market entry by months or even years. Some manufacturers push back. They argue the tests are too rigid. But the FDA’s position is clear: dissolution must be product-specific. There’s no universal formula. Every drug has its own story - and its own test.The Bigger Picture
Dissolution testing is part of a larger system that keeps our drug supply safe. It’s connected to other FDA tools: the Biopharmaceutics Classification System (BCS), the Dissolution Methods Database, and the ANDA review process. Together, they form a web of checks that prevent unsafe or ineffective drugs from reaching patients. The FDA continues to refine this system. In 2022, they began exploring biowaivers for BCS Class III drugs - those with high solubility but low permeability. If approved, this could expand the use of dissolution testing even further. But the core principle remains unchanged: if you can’t prove a generic drug releases its medicine the same way as the original - it doesn’t get approved. No exceptions. No compromises.What is dissolution testing in generic drugs?
Dissolution testing is a laboratory procedure that measures how quickly a drug releases its active ingredient in a simulated environment, like stomach fluid. For generic drugs, it proves the product releases medicine at the same rate and amount as the brand-name version, ensuring they work the same way in the body.
Why doesn’t the FDA require human studies for every generic drug?
The FDA uses dissolution testing as a scientifically validated substitute for human bioequivalence studies. When a generic drug’s dissolution profile matches the brand-name drug - especially for high-solubility drugs (BCS Class I) - the agency can confidently approve it without testing on people. This saves time, money, and resources while maintaining safety.
What is the f2 similarity factor?
The f2 similarity factor is a mathematical tool used by the FDA to compare the dissolution profiles of two drug products. An f2 score of 50 or higher means the two profiles are statistically similar. A score below 50 indicates a significant difference and usually leads to rejection of the generic application.
Do all generic drugs need dissolution testing?
Yes - all oral solid dosage forms (tablets, capsules) and oral suspensions must undergo dissolution testing. Oral liquids and topical products are generally exempt because they’re already in solution. The test is mandatory under the Abbreviated New Drug Application (ANDA) process.
How does the FDA handle modified-release drugs?
For extended-release products, the FDA requires testing under multiple pH levels (1.2, 4.5, and 6.8) to simulate different parts of the digestive tract. They also perform alcohol challenge tests to check for dose-dumping risks. These drugs must show consistent release under all conditions before approval.
Can a generic drug be approved even if its dissolution profile differs from the brand?
Rarely. If dissolution data shows a significant difference but human bioequivalence studies prove the drug works, the FDA may still approve it - but only after demanding additional justification. In most cases, the generic must match the brand’s profile exactly, or it’s rejected.



Gabrielle Conroy
February 24, 2026 AT 02:45Christopher Wiedenhaupt
February 25, 2026 AT 06:53John Smith
February 26, 2026 AT 08:06Shalini Gautam
February 26, 2026 AT 15:00Natanya Green
February 27, 2026 AT 10:19Steven Pam
February 27, 2026 AT 11:19Timothy Haroutunian
March 1, 2026 AT 09:18Erin Pinheiro
March 3, 2026 AT 07:38Michael FItzpatrick
March 3, 2026 AT 20:59Brandice Valentino
March 3, 2026 AT 21:30Larry Zerpa
March 4, 2026 AT 00:45Nandini Wagh
March 5, 2026 AT 08:00Ashley Johnson
March 6, 2026 AT 05:05tia novialiswati
March 6, 2026 AT 16:24Lillian Knezek
March 7, 2026 AT 20:14